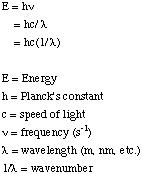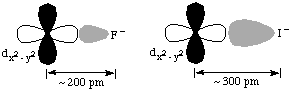![]()
B. The Spectrochemical Series
C. Magnitudes of the Octahedral Splitting Energy
D. Crystal Field Stabilization Energy (CFSE) in Octahedral Complexes
A. Absorption Spectra and Colours of Complexes
The visible spectrum is shown below, with colours that correspond to the wavelength of the light. "White light" is a combination of all of these colours. When a solution or coloured glass absorbs part of the visible spectrum, the colour that is seen is due to the remainder of the spectrum. If there is just a single absorption peak in the visible spectrum of a solution, the colour seen is the complementary colour.
Below the visible spectrum are the corresponding complementary colours. Thus, if a solution absorbs red light, its colour will be blue-green. If the solution absorbs green light, its colour will be purple.
![]()
It is common in transition metal chemistry to report the crystal field splitting energy, the pairing energy, and even the absorption spectra in terms of the wavenumber rather than the wavelength. The wavenumber is the number of waves in a given length. The relationships between the frequency, the wavelength, the wavenumber, and the energy are as follows:
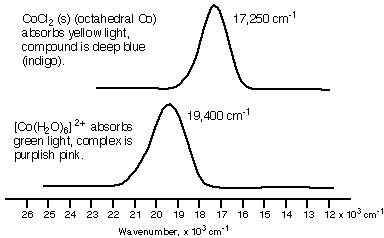
Now we will look at the absorption spectra of two cobalt(II) species. Crystalline cobalt(II) chloride contains octahedral cobalt(II) surrounded by six chloride ions. It is deep blue in colour, because the cobalt absorbs light in the yellow part of the spectrum. On the other hand, the hexaaquocobalt(II) cation absorbs light in the green part of the spectrum, resulting in a purplish pink colour.
B. The Spectrochemical Series
Recall that the reason these two high-spin cobalt species are coloured is because the energy separation between the t2g and the eg levels in the cobalt (II) ion lies in the visible region of the spectrum. Thus absorption of light occurs because an electron can be excited from a lower energy level (a t2g orbital in this case) to a higher energy orbital (eg in this case).
excitation.gif)
The reason that the two species are differently colored is because the d-orbital splitting energy is different when chloride is a ligand than when water is a ligand.
It is possible to arrange ligands into a series that reflects their ability to split the d-orbitals. This series is essentially the same no matter what the metal ion is. Thus, water not only splits the d-orbitals more than chloride for cobalt(II), but also for cobalt(III), iron(II), iron(III), nickel(II), platinum(IV), chromium(III), and so on. This series is as follows:
The positions of some of these ligands can be explained, or at least the ligands can be classified according to their donor/acceptor properties. Three groupings are considered in the following paragraphs.I- < Br- < Cl- < SCN- < NO3- < F- < OH- < H2O < NCS- < gly < py < NH3 < en < NO2- < PPh3 < CN- < CO
1. I- < Br- < Cl- < F-. The crystal field model looks at electrostatic repulsions between the ligands and electrons in d-orbitals on the metal ion. The smaller the ligand, the closer it comes to the metal ion and thus the greater the repulsion.C. Magnitudes of the Octahedral Splitting Energy
2. F- < OH- < H2O. Fluoride and hydroxide lie below water in the spectrochemical series because they are both pi-donor ligands. That is, F- and OH- can rehybridize and donate a pair of electrons from their p-orbitals to d-orbitals on the metal, forming a pi-bond as shown below. This reduces the negative charge on the fluoride and the positive charge on the metal, so Deltao is reduced.
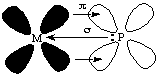
3. PPh3 < CN- < CO. It is surely astonishing to many chemists that not only do carbon monoxide and phosphine ligands bond readily to many transition metals, but that of all the ligands, they (together with cyanide) have the greatest capacity to split the d-orbitals. Let's consider what happens when a bond is formed between a metal ion and a phosphine ligand. The bond distance is relatively large (larger than the M-N distance in ammine complexes), so one would expect phosphines to fall lower in the spectrochemical series, as observed in the iodide-bromide-chloride-fluoride series.
If the metal ion has electrons in its d-orbitals, it can donate them to the phosphine ligand through the empty d-orbitals on phosphorus:
Cyanide and carbon monoxide behave similarly to phosphine ligands, but they make use of their empty anti-bonding pi-orbitals to accept electrons from the metal.
"Normal" bonding occurs when a ligand donates electrons to a metal. When a metal ion donates electrons back to the ligand, this is called back-bonding. The combination of normal bonding and back-bonding creates a strong bond between the ligand and the metal. The reason that phosphines, carbon monoxide, and cyanide are so poisonous is because they bond readily to iron in biological systems and cannot be displaced by the ligands (such as oxygen) which should bond to iron in normal metabolic processes.
The following table shows the magnitudes of the octahedral splitting energy as a function of the ligand.
There is a factor of 2 between the weakest and the strongest ligands. This will translate into a difference in the Crystal Field Stabilization Energy of nearly 200 kJ/mole, i.e. the hexacyanochromate(III) complex is more stable than the hexachlorochromate complex by 200 kJ/mole due to d-orbital splitting.
Complex [CrCl6]3- 13,200 [Cr(H2O)6]3+ 17,400 [Cr(NH3)6]3+ 21,500 [Cr(en)6]3+ 21,900 [Cr(CN)6]3- 26,600
The next table shows typical variations in the octahedral splitting energy with changing oxidation state of the metal and changing period number. (Cobalt, rhodium, and iridium are in the same group, but belong to different periods).
It is clear that the octahedral splitting energy increases significantly with the oxidation state of the metal, even when the ligands are the same. Another important observation is that the octahedral splitting energy increases as the central metal ion moves from one period to another (down a column) in the periodic table. This is the principal reason that nickel(II) complexes behave quite differently from platinum(II) complexes.
Complex [Co(NH3)6]2+ 10,100 [Co(NH3)6]3+ 22,900 [Rh(NH3)6]3+ 34,100 [Ir(NH3)6]3+ 41,000
D. Crystal Field Stabilization Energy (CFSE) in Octahedral Complexes
The crystal field stabilization energy is defined as the energy by which a complex is stabilized (compared to the free ion) due to the splitting of the d-orbitals. It is easily calculated:
![]()
The Pairing Energy correction is necessary only when the complex (low-spin) has fewer unpaired electrons than the free ion. The Pairing Energy is positive in the thermodynamic sense, i.e. pairing is an endothermic process because energy is required to make it happen.
Example. Let's calculate the crystal field Stabilization Energy for high-spin and low-spin octahedral cobalt(III) in terms of the splitting energy and the pairing energy.Solution. Cobalt(III) is a d6 metal ion. The diagrams for the free ion, high-spin cobalt(III), and low-spin cobalt(III) are shown below:
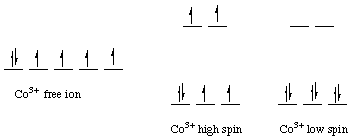
The CFSE for the high-spin case isCFSEhigh-spin = (4 electrons) (-2/5 Deltao) + (2 electrons)(+3/5 Deltao)The CFSE for the low-spin case is= -0.4 Deltao
CFSElow-spin = (6 electrons) (-2/5 Deltao) + (0 electrons)(+3/5 Deltao) + 2PNote that the pairing correction is 2 P and not 3 P. There is already one electron pair in free Co(III), and in low-spin Co(III) there are three electron pairs. The difference is 2 pairs of electrons, or 2 P in the CFSE calculation.= -2.4 Deltao + 2 P
Below is a table showing the crystal field stabilization energies for all d-electron configurations in an octahedral crystal field. You should be able to calculate these values without reference to the table.
Crystal Field Stabilization Energies for Octahedral Complexes of d1 - d10 Ions
# d-electrons CFSE high-spin CFSE low-spin 1 - 0.4 Deltao 2 - 0.8 Deltao 3 - 1.2 Deltao 4 - 0.6 Deltao - 1.6 Deltao + P 5 zero - 2.0 Deltao + 2 P 6 - 0.4 Deltao - 2.4 Deltao + 2 P 7 - 0.8 Deltao - 1.8 Deltao + P 8 - 1.2 Deltao 9 - 0.6 Deltao 10 zero
This page is http://chemiris.labs.brocku.ca/~chemweb/courses/chem232/CHEM2P32_Lecture_10.html
Created January 22, 2001 by M. F. Richardson
© Brock University, 2001