Lecture 12
Further Consequences of d-Orbital Splitting
A. Factors Determining the Stability of a Complex
B. Preferred Geometries: Octahedral Site Preference Energy (OSPE)
C. Preferred Geometries: Why Square Planar Nickel and Platinum?
D. Variations in Reactivity
A. Factors Determining the Stability of a Metal Complex
1. Number of ligand atoms coordinated to the metal (the
coordination number). Other things being equal, the more ligands that
are coordinated, the greater the bond energy. Thus 6-coordinate complexes
are inherently more stable than 4-coordinate complexes, but not by the
ratio of 6:4 because of the next factor.
2. Bond energy for each M-L bond. The greater the bond energy,
the more stable the complex. This factor interacts with the first. Four-coordinate
complexes have shorter bonds than six-coordinate complexes, and shorter
bonds are stronger bonds. For a divalent ion such as Ni(II), the estimated
bond energy for each Ni-OH2 bond in [Ni(H2O)6]2+
is about 300 kJ/mol; for the tetraaquo complex in either tetrahedral or
square geometry it is about 350 kJ/mol. [Estimated from hydration energy
for the Ni(II) ion.]
3. Inherent nature of the ligand. Chelating ligands are an example
here; complexes of polydentate ligands are more stable than corresponding
monodentate complexes (e.g. octahedral [Ni(en)3]2+
is more stable than octahedral [Ni(NH3)6]2+).
But there are other, more subtle examples. For example, complexes of NH3
are more stable than complexes of N-bonded NCS-. And certain
metal ions prefer specific donor atoms over others, which we will learn
more about later on in the course when we study Hard-Soft Acid-Base
Theory.
4. The crystal field stabilization energy (CFSE). It is possible
to calculate CFSE's in any geometry in terms of the octahedral splitting
energy Deltao; we've shown how to do it for three common geometries:
octahedral, tetrahedral, and square. Clearly, the CFSE is important, and
it depends on the number of d-electrons, and which orbitals they occupy.
However, it's not just the CFSE in terms of Deltao that
we need, it's the value in kJ/mol, and that depends on the actual magnitude
of Deltao for a given complex. Remember that Deltao
depends on these factors:
-
The specific metal ion. Even if two metal ions have the same charge
and the same ligands, and are in the same period of the periodic table,
they can have different values of Deltao. Some examples:
|
Metal Complex
|
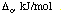
|
|
[Cr(H2O)6]2+
|
166
|
|
[Mn(H2O)6]2+
|
93
|
|
[Fe(H2O)6]2+
|
124
|
|
[Ni(H2O)6]2+
|
111
|
-
The charge on the metal ion. We've seen in a previous lecture that
the charge on the metal ion affects the value of Deltao for
a given set of ligands.
|
Complex
|
|
|
[Co(NH3)6]2+
|
121
|
|
[Co(NH3)6]3+
|
274
|
-
The period number of the metal ion for metals in the same group.
We've also seen that the octahedral splitting increases by about 50% on
going from the 4th period to the 5th, and by another 25% on going from
the 5th period to the 6th.
|
Complex
|
|
|
[Co(NH3)6]3+
|
274
|
|
[Rh(NH3)6]3+
|
408
|
|
[Ir(NH3)6]3+
|
491
|
-
The nature of the ligand. This is just a restatement of the spectrochemical
series, that ligands can be arranged in order of their ability to split
the d-orbitals.
|
Complex
|
|
|
[CrCl6]3-
|
158
|
|
[Cr(H2O)6]3+
|
208
|
|
[Cr(NH3)6]3+
|
257
|
|
[Cr(en)6]3+
|
262
|
|
[Cr(CN)6]3-
|
318
|
B. Preferred Geometries: The preference for octahedral coordination
over tetrahedral coordination and the Octahedral Site Preference Energy
(OSPE)
In Lecture 8 we raised several questions that we wanted a bonding theory
to explain. One of them had to do with the pronounced preference of some
metal ions for a specific coordination number and geometry. For example:
-
Cr3+, Co3+, and Pt4+ were cited as ions
that are found almost exclusively in octahedral coordination.
-
On the other hand, Pt2+ was stated to be an ion that occurred
only in square planar coordination. Other ions that occur only in square
planar coordination include Au3+, Rh+, and Ir+.
-
A few metal ions (high-spin Fe3+, high-spin Mn2+,
and Zn2+ among them) seem to have a fair proportion of tetrahedral
complexes in addition to octahedral complexes, but no square planar complexes.
Let's begin by looking at the electronic configurations of the ions just
mentioned.
|
Metal ions and geometries
|
# of d-electrons
|
d-orbital diagram
|
CFSE (neglect P)
|
|
Cr3+ (octahedral)
|
3
|
t2g3
|
-1.2 Deltao
|
|
low-spin Fe2+, Co3+, Pt4+ (octahedral)
|
6
|
t2g6
|
-2.4 Deltao
|
|
Pt2+, Au3+, Rh+ (square)
|
8
|
dxz2dyz2dz22dxy2
|
-2.44 Deltao
|
|
high-spin Fe3+, Mn2+ (octahedral/tetrahedral)
|
5
|
t2g3eg2 (oct); e2t23
(tet)
|
zero whether octahedral or tetrahedral
|
|
Zn2+ (octahedral)
|
10
|
t2g6eg4 (oct); e4t26
(tet)
|
zero whether octahedral or tetrahedral
|
There is a correlation between electronic configuration and preferred
geometries. Ions from different groups and different periods in the periodic
table have similar properties based on the number of d-electrons and the
crystal field stabilization energies.
1. The octahedral site preference energy (OSPE) for Co(III)
and other low-spin d6 ions is very large. The CFSE can be
calculated for octahedral and tetrahedral cobalt(III) complexes as shown
in the Figure below. The octahedral complex is much more stabilized by
the d-orbital splittings than the tetrahedral complex. This

The difference between the crystal field stabilization energies for
an octahedral complex and a complex in another geometry (tetrahedral in
this case) is called the octahedral site preference energy. The
actual magnitude of the difference above, -2.13 Deltao, is several
hundred kilojoules since the octahedral splitting energy varies from about
60 kJ for the weakest ligands to more than 300 kJ for the strongest.
Thus, not only is octahedral coordination for Co(III) favored by the
larger number of bonds compared to tetrahedral complexes, but also by the
CFSE.
2. The octahedral site preference energy (compared to tetrahedral
coordination) is also very large for Cr(III) and other d3 ions,
and for low-spin complexes of d4, d5, and d7
ions.
The graph below gives the OSPE in terms of Deltao for every
d-electron configuration. Again, octahedral coordination is not only favored
by the greater number of bonds compared to tetrahedral, but also by the
greater crystal field stabilization energy for octahedral d3
and low-spin d4, d5, and d7 complexes.
It is not surprising that there are so many octahedral complexes for these
ions.
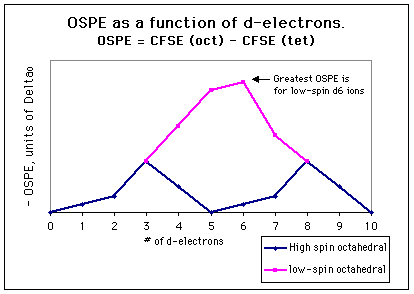
3. Some metal ions have no electronic preference for octahedral coordination
over tetrahedral coordination. For high-spin d5 complexes,
d0 complexes, and d10 complexes, there is no difference
in the crystal field stabilization energies in octahedral and tetrahedral
geometries; they are zero in both geometries. The OSPE is small even for
d1, d2, and high-spin d6 and d7
ions. Ions with these d-electron configurations are the most likely to
be associated with tetrahedral complexes, as factors other than electronic
structure determine the preferred geometry. Among the d0 ions
are Ti(IV), V(V), and Mo(VI); the most common d5 ions are Mn(II)
and Fe(III); and Zn(II), Cd(II), Hg(II), Cu(I), Ag(I), and Au(I) are the
most common d10 ions. Many tetrahedral complexes are known for
these ions.
C. Nickel(II) vs. platinum(II): why is Pt2+ found only in
square planar coordination but Ni2+ is found in several geometries?
Let's consider a single example. Nickel forms the octahedral
hexaammine complex, [Ni(NH3)6]2+, but
platinum forms the square planar tetraammine complex [Pt(NH3)4]2+.
Both metal ions have 8 d-electrons. We can calculate a preference energy
for octahedral over square planar coordination, just as we did for octahedral
over tetrahedral. We will neglect the pairing energy for this calculation.
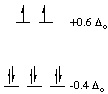
|
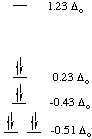
|
|
CFSEoct = -1.2 Deltao
|
CFSEsq.pl. = -2.44 Deltao
|
Notice that in this case, the crystal field stabilization (in terms of
Deltao) is much greater for square planar coordination than
for octahedral coordination! In fact, the preference energy (compared to
octahedral coordination) for square planar coordination of a d8 metal ion
is -1.24 Deltao. However, let's use the following values and
determine the absolute values for the square planar preference in kJ/mol.
|
Complex
|
|
|
[Ni(NH3)x]2+
|
132
|
|
[Pt(NH3)x]2+
|
359
|
Preference for square planar coordination by [Ni(NH3)x]2+
= -1.24 (132 kJ/mol)
= -164 kJ/mol
Preference for square planar coordination by [Pt(NH3)x]2+
= -1.24 (359 kJ/mol)
= -445 kJ/mol
Thus square planar [Pt(NH3)4]2+ is stabilized
by nearly 300 kJ/mol more than the corresponding nickel complex. This greater
energy difference is enough to make up for the loss in bond energy suffered
as an octahedral platinum(II) complex is transformed to a square planar
complex, but is not usually enough to favor square planar complexes for
nickel.
So why does nickel form square planar complexes at all? Actually, there
are many fewer square planar complexes than there are octahedral ones.
the ones that are square planar tend to be found with stronger-field ligands,
i.e. those that are capable of giving high values for the octahedral splitting
energy Deltao.
D. Variations in Reactivity.
Let's take as a single example the difference in ligand substitution
rates for the complexes [Co(NH3)6]3+ and
[Ni(NH3)6]2+. Each complex has a negative
enthalpy for the following reaction, so both complexes are thermodynamically
capable of undergoing ligand exchange with water.
[M(NH3)6]n+ + 6 H3O+
---> [M(H2O)6]n+ + 6 NH4+
However, the exchange reaction for the nickel complex is very fast,
while the reaction for the cobalt complex takes days or weeks to go to
completion. Why is this?
The short answer is found if we consider the reaction mechanism, i.e.,
how the reaction must take place. There are two limiting possibilities,
both of them involving a 2-step reaction as shown below.
Mechanism 1. One of the NH3 ligands dissociates from
the complex to give a 5-coordinate intermediate. This intermediate then
forms a complex with a water molecule.
[M(NH3)6]n+ ---> [M(NH3)5]n+
+ NH3
[M(NH3)5]n+ + H2O --->
[M(NH3)5H2O]n+
Repetition of this process ultimately gives the hexaaquo ion.
Mechanism 2. A water molecule coordinates to the complex to give
a 7-coordinate intermediate. This intermediate then loses an ammonia molecule.
[M(NH3)6]n+ + H2O --->
[M(NH3)6H2O]n+
[M(NH3)6H2O]n+ ---> [M(NH3)5H2O]n+
+ NH3
The process repeats itself until the hexaaquo ion is formed.
Each of these mechanisms involves a reaction to give a reactive intermediate
with a different coordination number (5 or 7).
When M = Co(III) (low-spin d6), considerable crystal field
stabilization is lost because the CFSE is so much greater for octahedral
coordination than the CFSE than any possible geometry in 5- or 7-coordination.
The loss in CFSE on going from 6-coordination to 5- or 7-coordination
increases the activation energy for the reaction, thus slowing it.

For most other ions, there is less loss (or even a gain) in CFSE on
going from 6- to 5- or 7-coordinate intermediates. Thus exchange reactions
for these ions are not retarded by the loss in CFSE.
Back
to Lecture Schedule
Back
to CHEM 2P32 Home Page
This page is http://chemiris.labs.brocku.ca/~chemweb/courses/chem232/CHEM2P32_Lecture_12.html
Created February 2, 2001 by M. F. Richardson.
© Brock University, 2001